Structural Features of Protein (Motif, Domain)
Proteins are the workhorses of the cell, performing a vast array of functions. Their abilities are intricately linked to their three-dimensional structures. In this lesson, we’ll delve into the fascinating world of protein architecture, focusing on key structural features known as motifs and domains. Understanding these concepts is fundamental to fields like protein modeling, drug design, and molecular biology.
Learning Objectives:
- Understand the basics of protein modeling and its significance.
- Describe the four levels of protein structural hierarchy.
- Explain how proteins are classified based on their structure.
- Clearly define and differentiate between protein motifs and domains.
- Recognize examples of common motifs and domains.
Let’s begin by understanding the broader context of how we study these intricate structures.
1. What is Protein Modeling? (And Why Should We Care?)
Protein modeling is the process of predicting or deducing the three-dimensional structure of a protein from its amino acid sequence, or refining an existing structure. It’s like being a molecular architect!
There are three main aspects to protein modeling:
-
Structure Prediction:
- What? Starting with just the amino acid sequence (like a string of letters), scientists use computational methods to predict its 3D folded shape. This is crucial when experimental structures (from X-ray crystallography or NMR) are unavailable.
- Analogy: Imagine trying to build a complex 3D puzzle (the protein structure) when you only have the list of pieces (the amino acid sequence).
-
Structure Refinement:
- What? Improving the accuracy of an existing, often experimentally determined or roughly modeled, protein structure. This can involve fixing errors, adding missing residues, or optimizing the geometry.
- Analogy: Taking a slightly misshapen clay sculpture and carefully smoothing out imperfections or correcting proportions.
-
Protein Dynamics:
- What? Studying how proteins move, flex, and change shape. Proteins are not static; their movements are often essential for their function.
- Analogy: Understanding not just the static blueprint of a machine, but how its gears and levers move and interact.
Why is Protein Modeling Important?
Understanding protein structure is key to understanding function. Protein modeling has profound implications:
- Drug Discovery: Designing drugs that bind specifically to target proteins (e.g., enzymes in a pathogen, receptors on a cancer cell). If you know the lock’s shape (protein), you can design a better key (drug).
- Enzyme Engineering: Modifying enzymes for industrial applications (e.g., making laundry detergents more effective, or creating enzymes for biofuel production).
- Structural Biology: Understanding diseases caused by misfolded proteins (e.g., Alzheimer’s, Parkinson’s, prion diseases). It also helps in classifying proteins into families.
- Systems Biology: Deciphering how proteins interact within complex cellular networks and pathways, like signaling pathways or immune responses.
2. The Building Blocks: Structural Hierarchy of Proteins
Proteins exhibit a hierarchical organization of structure, typically described in four levels.
2.1 Primary Structure
- Definition: The linear sequence of amino acids in a polypeptide chain, linked by peptide bonds.
- Analogy: The specific order of letters that make up a word.
P-R-O-T-E-I-Nis different fromT-R-O-P-E-I-N. - This sequence is determined by the gene encoding the protein.
- It dictates all higher levels of structure.
2.2 Secondary Structure
- Definition: Local, regularly repeating structures formed by hydrogen bonding between atoms of the polypeptide backbone (not the R-groups).
- Analogy: Common spelling patterns or short, recognizable phrases within a sentence, like “ing” or “tion”.
- The two most common types are:
- α-helix (Alpha-helix): A right-handed coil or spiral structure, where the backbone N-H group of one amino acid forms a hydrogen bond with the backbone C=O group of an amino acid four residues earlier.
- Think of a spiral staircase or a coiled ribbon.
- β-sheet (Beta-pleated sheet): Formed when two or more segments of a polypeptide chain (β-strands) line up next to each other, and hydrogen bonds form between them. They can be parallel (strands run in the same direction) or antiparallel (strands run in opposite directions).
- Think of a sheet of paper folded into pleats. Loops and Turns: These are non-regular secondary structures that connect α-helices and β-sheets, allowing the polypeptide chain to change direction.
- α-helix (Alpha-helix): A right-handed coil or spiral structure, where the backbone N-H group of one amino acid forms a hydrogen bond with the backbone C=O group of an amino acid four residues earlier.
2.3 Tertiary Structure
- Definition: The overall three-dimensional shape of a single polypeptide chain. It arises from interactions between the R-groups (side chains) of the amino acids.
- Analogy: The complete 3D structure of a complex origami figure, formed by specific folds (secondary structures) and further interactions (R-group interactions).
- Types of R-group interactions stabilizing tertiary structure:
- Hydrophobic Interactions: Nonpolar R-groups tend to cluster together in the interior of the protein, away from water.
- Ionic Bonds: Attractions between positively and negatively charged R-groups.
- Hydrogen Bonds: Form between polar R-groups.
- Disulfide Bridges: Strong covalent bonds formed between the sulfur atoms of two cysteine residues.
- Van der Waals Interactions: Weak, short-range attractions between all atoms.
2.4 Quaternary Structure
- Definition: The arrangement of multiple polypeptide chains (subunits) to form a functional protein complex. Not all proteins have quaternary structure (many function as single polypeptides).
- Analogy: A team of individual players (polypeptide subunits) working together to form a functional unit (the protein complex), like a basketball team.
- The same types of interactions that stabilize tertiary structure also hold subunits together in quaternary structure.
- Example: Hemoglobin, which consists of four polypeptide subunits (two alpha and two beta). (Refer back to the Quaternary part of the image from slide 10).
3. Organizing the Structures: Protein Classification
With millions of protein structures known, how do scientists make sense of them all? They classify them! Protein structures can be grouped based on:
- Structural similarity: Overall shape and arrangement of secondary structural elements.
- Similar topology: The way secondary structures are connected.
- A common evolutionary origin: Implying they evolved from a common ancestor.
3.1 Structural Similarity (Class)
One common way to classify proteins is by their dominant secondary structure content:
- All α: Consist almost entirely of α-helices.
- All β: Consist almost entirely of β-sheets.
- α/β: Contain both α-helices and β-sheets that are largely interspersed or alternate. β-strands are usually parallel. Examples include TIM barrels and Rossmann folds.
- α+β: Contain both α-helices and β-sheets, but they are largely segregated into distinct regions. β-strands are often antiparallel.
3.2 Topology (Fold)
- Definition: The specific arrangement and connectivity of secondary structural elements (SSEs) within a protein domain. It describes the path of the polypeptide chain.
- Analogy: Different ways to arrange a set of LEGO bricks (SSEs) and connect them to build distinct, recognizable shapes (folds).
- Proteins with the same fold have the same major SSEs in the same arrangement and with the same topological connections.
- There are a limited number of common folds (~1000-1500) despite the vast number of different proteins.
Examples of Common Folds:
-
TIM Barrel (Triosephosphate Isomerase Barrel):
- An (α/β) fold, typically consisting of eight parallel β-strands forming a central barrel, surrounded by eight α-helices on the outside.
- A very common fold, often found in enzymes.
-
Rossmann Fold:
- Another α/β fold, characterized by a repeating βαβαβ motif (a β-strand, followed by an α-helix, then another β-strand, etc.).
- Often involved in binding nucleotides (like NAD⁺ or ATP).
The terms Fold, Supersecondary Structure, and Motif are related and sometimes used interchangeably, which can be confusing. We’ll clarify “Motif” in more detail in the next section. Slide 22 shows examples of different folds, also referred to as supersecondary structures or motifs in a broader sense.
3.3 Protein Structure Databases
Several databases classify protein structures hierarchically:
- SCOP (Structural Classification of Proteins):
- Levels: Class, Fold, Superfamily (evolutionary relationship), Family (closer evolutionary relationship).
- SCOPe: https://scop.berkeley.edu/
- SCOP2: https://scop.mrc-lmb.cam.ac.uk/
- CATH (Class, Architecture, Topology, Homologous superfamily):
- Class: Secondary structure content.
- Architecture: Overall shape and packing of secondary structures.
- Topology: The fold, describing connectivity.
- Homologous Superfamily: Groups proteins with a common ancestor.
- CATH: https://www.cathdb.info/
- ECOD (Evolutionary Classification of Protein Domains):
4. Key Architectural Elements: Motifs and Domains
Now we arrive at the core of our lesson: understanding protein motifs and domains. These are crucial for understanding protein structure-function relationships.
4.1 Protein Motifs (Supersecondary Structures)
- Definition: A motif, also known as a supersecondary structure or sometimes a (small) fold, is a recognizable, recurring, small combination of a few secondary structure elements (α-helices and/or β-strands) and the loops connecting them.
- Characteristics:
- They are “patterns” of local 3D structure.
- Often associated with a particular function (e.g., DNA binding, ion binding), but not always.
- Usually too small to fold independently and remain stable on their own; they are typically part of a larger domain.
- Analogy: A common musical riff or a specific type of knot – a simple, recognizable pattern. Or, specific short phrases or idioms in language (e.g., “helix-turn-helix”).
Common Structural Motifs:
- Helix-turn-helix (HTH): Two α-helices connected by a short turn. Often found in DNA-binding proteins.
- β-hairpin: Two antiparallel β-strands connected by a short loop (2-5 residues).
- Greek key motif: Four or more antiparallel β-strands arranged in a pattern resembling a Greek key design.
- β-α-β motif: Two parallel β-strands linked by an α-helix that sits above or below the plane of the strands. This is a fundamental unit of many α/β proteins like the Rossmann fold and TIM barrel.
- Zinc finger: A small motif where one or more zinc ions are coordinated by cysteine and/or histidine residues, stabilizing a small loop or finger-like projection. Often involved in DNA/RNA binding or protein-protein interactions.
Sequence Motifs vs. Structural Motifs:
- Sequence Motif: A short, conserved amino acid sequence pattern (e.g.,
N-X-S/Tfor N-glycosylation). It may or may not correspond to a defined structural motif but often implies a functional site. - Structural Motif: A conserved 3D arrangement of secondary structures, as described above.
4.2 Protein Domains
- Definition: A domain is a compact, stable, independently folding structural unit within a polypeptide chain. It’s a conserved part of a given protein sequence and (tertiary) structure that can evolve, function, and exist independently of the rest of the protein chain.
- Characteristics:
- Typically larger than motifs (usually 50-300 amino acids).
- The fundamental unit of protein evolution and function.
- Often associated with a specific function (e.g., a kinase domain catalyzes phosphorylation, an SH2 domain binds phosphotyrosine).
- Proteins can be single-domain or multi-domain. Multi-domain proteins are like a string of beads, where each bead is a domain.
- Analogy: A room in a house (the protein). Each room (domain) has its own structure and purpose (e.g., kitchen, bedroom) and can exist somewhat independently, but is part of the larger house. Or, different functional modules in a piece of software, each performing a specific task.
Relationship between Motif, Domain, and Fold:
- A domain is typically composed of one or more motifs and other secondary structural elements.
- These elements are arranged in a specific 3D structure that defines the domain’s fold.
- So, a domain has a particular fold, and that fold may be built up from recognizable motifs.
Example: SARS-CoV-2 Spike Protein Domains
Let’s look at a real-world example. The Spike (S) protein of SARS-CoV-2 (the virus causing COVID-19) is a multi-domain protein crucial for viral entry into host cells.
The Spike protein has several distinct domains:
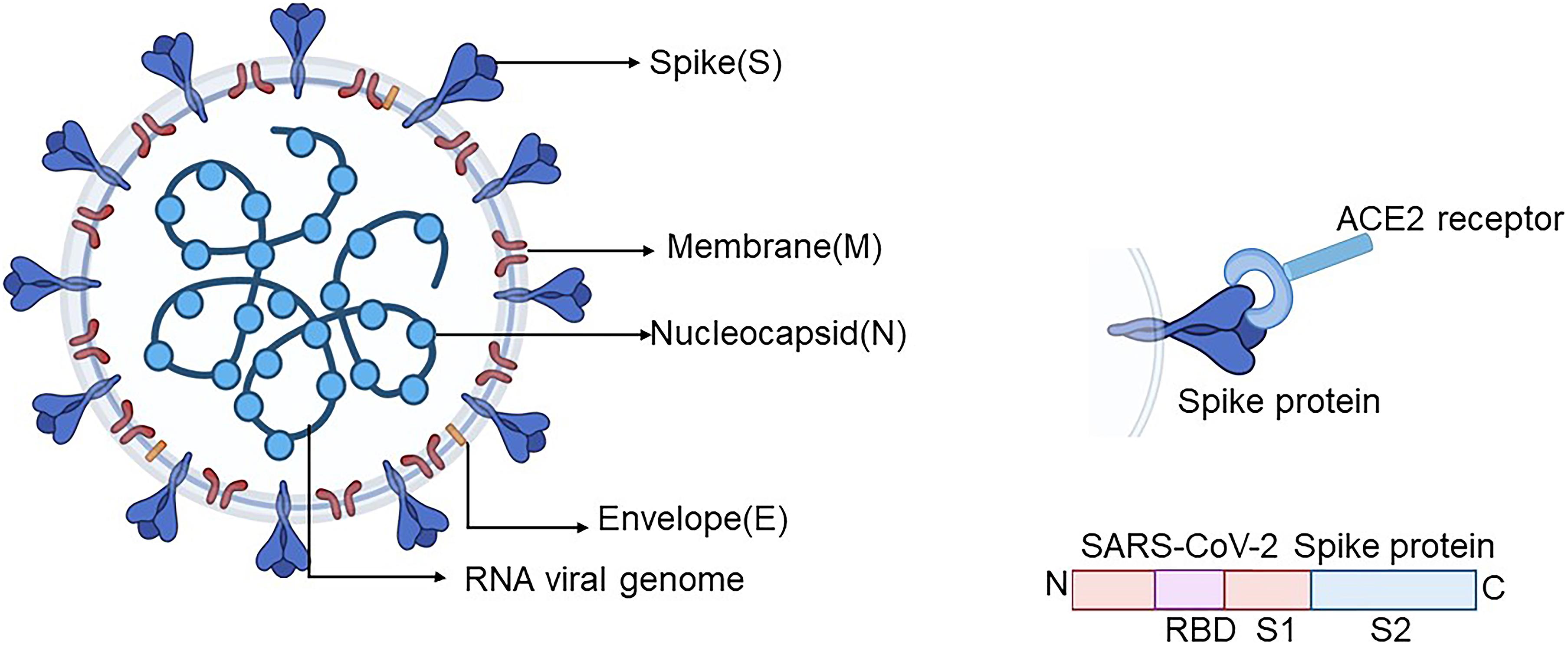
- S1 Subunit: Contains domains responsible for binding to the host cell receptor.
- NTD (N-Terminal Domain)
- RBD (Receptor Binding Domain): This is a critical domain that directly interacts with the ACE2 receptor on human cells.
- S2 Subunit: Contains domains responsible for membrane fusion.
How to Identify Motifs and Domains?
- Motifs:
- Sequence motifs can be found using databases like PROSITE.
- Structural motifs are identified by visual inspection of 3D structures or by computational programs that search for known patterns.
- Domains:
- Can often be identified by limited proteolysis (domains are often resistant to cleavage within their core).
- Sequence homology searches (e.g., using BLAST against domain databases).
- Databases like Pfam, SMART, InterPro, and CDD (Conserved Domain Database from NCBI) curate collections of protein domains and families.
4.3 Motif RegEx in Bioinformatics: Pattern Matching in Sequences
In bioinformatics, identifying specific patterns or sequence motifs within DNA, RNA, or protein sequences is crucial for understanding function, regulation, and evolution. One powerful way to describe and search for these motifs is by using Regular Expressions (RegEx).
What are Regular Expressions (RegEx)?
A Regular Expression (often shortened to RegEx or regexp) is a sequence of characters that defines a search pattern. Think of it as a highly advanced “find” or “search and replace” tool that uses a special syntax to describe patterns in text (in our case, biological sequences).
Analogy: Imagine you’re searching a large library for books.
- A simple search might be for an exact title: “Introduction to Biology”.
- A RegEx search is like saying: “Find me all books whose titles start with ‘Intro’, have ‘Bio’ somewhere in the middle, and end with a year between 1990 and 2020.”
RegEx allows for this kind of flexible and powerful pattern matching.
Why Use RegEx for Biological Motifs?
Biological motifs are rarely perfectly conserved. For instance, a binding site for a protein might tolerate some variations in its amino acid sequence. RegEx is well-suited to handle this:
- Flexibility: Define patterns that include exact characters, variable characters (e.g., any amino acid), specific choices (e.g., a Lysine or Arginine), and variable spacing between elements.
- Conciseness: A relatively short RegEx can describe a complex sequence pattern.
- Computational Power: Efficient algorithms exist to search large sequence databases using RegEx patterns.
Common RegEx Syntax for Biological Sequences
While RegEx syntax can be extensive, here are some common elements used in bioinformatics. (Note: The exact implementation can vary slightly between programming languages or tools.)
| Syntax | Description | Example (Protein) | Example (Nucleotide) | Meaning |
|---|---|---|---|---|
A | Exact character match | M | G | Matches Methionine (M) or Guanine (G) |
. | Wildcard: Matches any single character (in some contexts, any valid residue) | A.G | T.A | A, any amino acid, G; or T, any nucleotide, A |
X | (Commonly) Matches any amino acid | AXG | N/A | A, any amino acid, G |
N | (Commonly) Matches any nucleotide (A, C, G, T/U) | N/A | ANG | A, any nucleotide, G |
[ABC] | Character Set: Matches any one character inside the brackets | [ST] | [AG] (or R) | Serine (S) or Threonine (T); or Adenine (A) or Guanine (G) (Purine) |
[^ABC] | Negated Character Set: Matches any character NOT in the brackets | [^P] | [^T] | Any amino acid except Proline (P); any nucleotide except Thymine (T) |
A* | Zero or more occurrences of A | G* | C* | Zero or more Glycines; zero or more Cytosines |
A+ | One or more occurrences of A | L+ | A+ | One or more Leucines; one or more Adenines |
A? | Zero or one occurrence of A | P? | T? | Zero or one Proline; zero or one Thymine |
A{n} | Exactly ‘n’ occurrences of A | A{3} | G{2} | Exactly three Alanines (AAA); exactly two Guanines (GG) |
A{n,} | ’n’ or more occurrences of A | W{2,} | C{3,} | Two or more Tryptophans; three or more Cytosines |
A{n,m} | Between ‘n’ and ‘m’ occurrences of A (inclusive) | M{1,3} | T{2,4} | One to three Methionines; two to four Thymines |
(ABC) | Grouping: Treats ‘ABC’ as a unit for quantifiers or captures the match | (VI){2} | (CG){3} | Matches VIVI; Matches CGCGCG |
A|B | Alternation (OR): Matches A or B | D|E | A|T | Aspartic acid (D) or Glutamic acid (E); Adenine (A) or Thymine (T) |
IUPAC Ambiguity Codes: For nucleotides, standard IUPAC codes are often used directly or within character sets:
R= A or G (puRine)Y= C or T (pYrimidine)S= G or C (Strong interaction - 3 H-bonds)W= A or T (Weak interaction - 2 H-bonds)K= G or T (Keto)M= A or C (aMino)B= C or G or T (not A)D= A or G or T (not C)H= A or C or T (not G)V= A or C or G (not T)N= A or C or G or T (aNy)
For amino acids:
X= any amino acidB= Asparagine or Aspartic acid (Asx)Z= Glutamine or Glutamic acid (Glx)J= Leucine or Isoleucine
Examples of Biological Motifs using RegEx
-
N-glycosylation site motif:
- This is a site where a carbohydrate can be attached to an Asparagine (N) residue.
- The consensus sequence is
N-X-SorN-X-T, where X can be any amino acid except Proline (P). - A common RegEx (simplified):
N[^P][ST]N: Matches an Asparagine.[^P]: Matches any amino acid that is NOT Proline.[ST]: Matches either Serine or Threonine.
- Note: The actual PROSITE pattern is
N-{P}-[ST]-{P}, where{P}means “not P”. This highlights that specific databases might have their own RegEx-like syntax.
-
A hypothetical protein kinase C phosphorylation site:
- Many kinases recognize specific patterns. Let’s say a kinase recognizes Serine or Threonine if it’s preceded by an Arginine (R) or Lysine (K) two positions upstream, and followed by a hydrophobic amino acid (L, I, V, M, F) one position downstream.
- RegEx:
[RK].{1}[ST][LIVMF][RK]: Arginine or Lysine..{1}: Any one amino acid (spacer). (Xcould also be used:[RK]X[ST][LIVMF])[ST]: Serine or Threonine (the phosphorylation site).[LIVMF]: A hydrophobic residue.
-
Restriction enzyme EcoRI recognition site (DNA):
- EcoRI cuts at
G A A T T C. - RegEx:
GAATTC(a simple exact match).
- EcoRI cuts at
Tools and Databases
- PROSITE: A database of protein families and domains. It’s famous for using its own well-defined syntax for describing sequence motifs, which is a specialized form of regular expression. (e.g.,
N-{P}-[ST]-{P}for N-glycosylation). - Programming Languages: Python (module
re), Perl, R, Java, etc., all have built-in RegEx engines that bioinformaticians use extensively for custom script development to find motifs in sequence files (like FASTA). - Command-line tools:
grep(with-Efor extended RegEx or-Pfor Perl-compatible RegEx) can search for patterns in text files, including sequence files. - Specialized Bioinformatics Software: Many sequence analysis packages (e.g., UGENE, Geneious) have tools for RegEx-based motif searching.
Limitations and Considerations
- Complexity: Complex biological patterns can lead to very complicated and hard-to-read RegEx.
- Strictly Sequence-Based: Standard RegEx patterns don’t inherently understand 3D structure or evolutionary relationships (though the motifs they define often correlate with these).
- Sensitivity vs. Specificity:
- A very general RegEx (e.g.,
A.G) might find too many matches (high sensitivity, low specificity), including many false positives. - A very specific RegEx (e.g.,
ACEGICKLMNPQRSTVWY) might miss valid biological variations of the motif (low sensitivity, high specificity). - Finding the right balance is key and often requires experimental validation.
- A very general RegEx (e.g.,
- Syntax Variations: While core concepts are similar, minor syntax differences can exist between different RegEx engines (e.g., POSIX RegEx vs. Perl-compatible RegEx).
Understanding RegEx is a valuable skill for any bioinformatician, allowing for precise and flexible searching of biological sequence data for meaningful patterns.
5. Conclusion
Understanding protein structural features like motifs and domains is essential for dissecting how proteins work.
- Primary structure dictates the higher levels.
- Secondary structures (α-helices, β-sheets) are local folding patterns.
- Tertiary structure is the overall 3D shape of a single polypeptide, stabilized by R-group interactions.
- Quaternary structure involves multiple polypeptide subunits.
- Motifs are small, recognizable combinations of secondary structures.
- Domains are larger, independently folding, functional units that are the building blocks of most proteins.
- Protein classification systems (like SCOP and CATH) help organize the vast world of protein structures based on these features.
This knowledge forms the bedrock for more advanced topics in bioinformatics, structural biology, and drug design. Keep exploring these fascinating molecular machines!
I hope this lesson helps you understand protein motifs and domains better!
- Resources
- API
- Sponsorships
- Open Source
- Company
- xOperon.com
- Our team
- Careers
- 2025 xOperon.com
- Privacy Policy
- Terms of Use
- Report Issues